Les syndromes CAPS
Les maladies – caractéristiques des caps
Le gène responsable CIAS1 ou NAPL3 ou NLRP3 a été identifié sur le chromosome 1. Il est responsable de la formation d’une protéine appelée cryopyrine qui est impliquée dans la régulation de l’inflammation.
L’ensemble FCAS+MWS+CINCA représente les 3 facettes cliniques de la gravité croissante d’une même maladie génétique appelée CAPS.
CAPS : Cryopyrine Associated Périodic Syndrome (Syndrome Périodique Associé à la Cryopyrine).
Archives
Syndrome de Muckle-Wells : La présence de surdité serait associée à une sévérité accrue
Syndrome de Muckle-Wells : essai de l’anakinra chez l’enfant et chez l’adulte
Les symptômes du FCAS
Familial Cold Auto-inflammatory Syndrome
Urticaire familiale au froid
Ce syndrome a été tout d’abord rapporté dans des familles d’Amérique du Nord et d’Europe, mais des cas sporadiques ont été rapportés ailleurs.
Le diagnostic est évoqué cliniquement, il peut être confirmé par l’identification d’une mutation du gène NALP3. Bien qu’aucun essai clinique n’ait été publié à ce jour, l’Anakinra, un antagoniste du récepteur IL-1, a un effet préventif sur la survenue des symptômes liés au froid.
Le gène NALP3 également connu sous le nom de CIAS s’exprime dans les leucocytes périphériques et les chondrocytes ; il code pour la cryopyrine. Des mutations de NALP3 ont également été identifiées dans le syndrome de Muckle Wells et le syndrome CINCA.
Plusieurs laboratoires en Europe et en Amérique du Nord effectuent des séquençages de NALP3.
*Auteur : Dr HM Hoffman (février 2005)*.
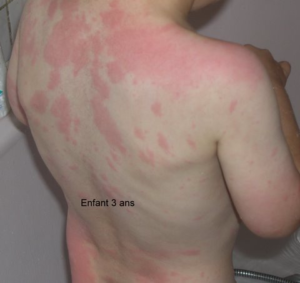
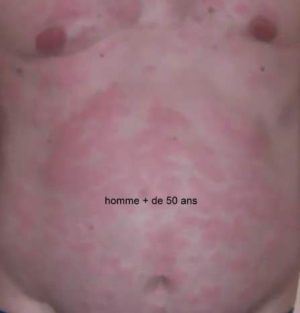
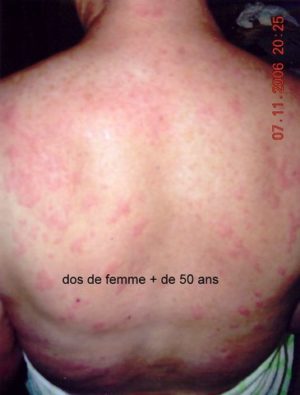
Les symptômes du MWS
Muckle et Wells Syndrome
Les premières manifestations sont une urticaire accompagnée d’une petite fièvre, non prurigineuse, parfois invalidante car presque permanente. Les autres signes inflammatoires sont essentiellement une atteinte articulaire (arthralgies ou arthrites), oculaire(conjonctivite). A ces signes s’associe une surdité neurosensorielle qui s’installe dans l’adolescence.
Les critères diagnostiques sont d’abord cliniques et maintenant assortis d’un diagnostic génétique. La transmission est autosomique dominante à expression variable intra et interfamiliale.
Le gène responsable CIAS1ou NAPL3 a été identifié sur le chromosome 1 en 1q44, il code pour une protéine appelée cryopyrine, qui appartient par son domaine N-terminal à la famille de la pyrine associée à la fièvre méditerranéenne familiale.
Des mutations du gène CIAS1 sont aussi associées à 2 autres entités : le Familial cold auto-inflammatory syndrome (FCAS) et le syndrome Chronic Infantile Neurological Cutaneous and Articular (CINCA). L’incidence est inconnue.
La surdité peut être appareillée.
* anakinra : dénomination commune internationale = nom commercial Kineret®
Les symptômes du CINCA
Chronique, Infantile, Neurologique, Cutané, Articulaire
Une centaine de cas seulement a été décrite dans le monde, mais ce syndrome est de mieux en mieux reconnu par les pédiatres.
L’éruption maculo-papuleuse de type urticarienne est très souvent présente dès la naissance et variable dans le temps. Elle est en réalité pseudo-urticarienne avec un infiltrat inflammatoire périvasculaire à polynucléaires.
La fièvre est inconstante et le plus souvent modeste.
L‘atteinte articulaire, d’expression variable, peut induire des poussées articulaires inflammatoires ponctuelles ne laissant pas d’anomalie entre les crises, ou de façon imprévisible, être responsable d’une hypertrophie non inflammatoire des cartilages de conjugaison et des épiphyses des os longs donnant un aspect pseudo-tumoral des articulations.
L’atteinte du système nerveux central se traduit par des céphalées chroniques en rapport avec une méningite chronique à polynucléaires.
Dans les formes sévères, un retard mental est possible.
L’atteinte neurosensorielle inclut une atteinte oculaire, de nature inflammatoire (uvéite, atteinte papillaire, névrite optique pouvant entraîner une cécité) et souvent une surdité de perception progressive. Ce syndrome évolue dans un contexte d’inflammation chronique. Il peut y avoir un retard de croissance, parfois une prématurité et une dysmaturité.
Une amyloïdose secondaire peut apparaître avec le temps. La biologie montre un syndrome inflammatoire non spécifique avec une anémie, une hyperleucocytose à polynucléaires, une augmentation de la vitesse de sédimentation et de la protéine réactive C (CRP), sans auto-anticorps ni déficit immunitaire.
Beaucoup de cas de CINCA sont sporadiques, mais des formes familiales à transmission autosomique dominante ont été décrites. Chez 60% des patients, une mutation du gène CIAS1 est mise en évidence, qu’il s’agisse de formes familiales ou sporadiques. Ce gène est fortement exprimé dans les cellules de l’immunité innée, en particulier les polynucléaires et les monocytes. Il code pour la cryopyrine qui joue un rôle important dans le contrôle de la réponse immune en particulier de l’immunité innée. Des mutations de ce gène sont également en cause dans le syndrome de Muckle-Wells et l’urticaire au froid, deux entités phénotypiquement proches du CINCA (voir ces termes).
Ces trois syndromes représentent un spectre de sévérité croissante d’une même pathologie :
l’urticaire familial au froid est la forme la plus bénigne,
le CINCA, la forme la plus sévère et le Muckle-Wells correspond à un phénotype de sévérité intermédiaire.
Le diagnostic est avant tout clinique, confirmé ensuite par la génétique. Le diagnostic différentiel inclut les pathologies auto-inflammatoires systémiques.
De nombreux médicaments anti-inflammatoires et immunosuppresseurs ont été essayés sans succès. Les corticoïdes améliorent la symptomatologie de façon incomplète et au prix d’une toxicité importante.
- L’anakinra – nom commercial « Kineret® », est un antagoniste du récepteur de l’interleukine-1 bêta.
- Le canakinumab – nom commercial « Ilaris® », est un anticorps monoclonal anti-IL-1. ayant obtenu l’AMM(autorisation de mise sur le marché) pour les CAPS.
Il existe une grande variabilité en terme de sévérité au sein de ce syndrome. Certains patients présentent le tableau clinique pathognomonique complet avec début précoce, atteinte du système nerveux central et arthropathies à début précoce. La qualité de vie de ces malades est souvent médiocre et le pronostic fonctionnel réservé du fait des rétractions tendineuses secondaires aux arthropathies. Certains patients présentent un phénotype clinique plus atténué et une meilleure qualité de vie.
*Auteur : Dr B. Neven (février 2007)*.
Les fièvres récurrentes héréditaires
- la fièvre méditerranéenne familiale (ou maladie périodique),
- le syndrome auto-inflammatoire associé au récepteur du TNF (ou TRAPS),
- les pathologies auto-inflammatoires systémiques associées à la Cryopyrine ou CAPS regroupant plusieurs maladies :
– l’urticaire familiale au froid,
– le syndrome de Muckle – Wells,
– le syndrome chronique infantile neurologique cutané et articulaire ou CINCA, - les déficits en mévalonate kinase regroupant :
– le syndrome hyper IgD,
– l’acidurie mévalonique, - les fièvres récurrentes auto-inflammatoires idiopathiques,
- le syndrome de Marshall dénommé également PFAPA.
Lien pour consulter cet ouvrage
- Page 10 : mode de transmission – les Caps sont de transmission autosomique dominante,
- Page 11 : le cas sporadique de la mutation ‘de novo’,
- Page 18 : les complications ‘l’amylose’ et la SAA,
- Page 19 : la nécessité d’un suivi régulier -prises de sang et analyses d’urine,
- Page 29 : les généralités sur les CAPS,
- Pages 39 à 34 : les syndromes, dont le MWS – 40 familles en France- et les traitements,
- Page 41 : les dates de découverte des gènes, c’est en 2001 que le gène CIAS1 a été découvert pour les CAPS,
- Page 42 : exemple de recherche : « Fièvres récurrentes héréditaires : étude fonctionnelle des gènes connus et clonage positionnel des gènes non encore identifiés » par le Pr Gilles Grateau, Paris, France. (Notre centre de référence pour adultes),
- Page 43 : les congrès, dont Amsterdam – voir mon compte rendu sur www.amws-cinca.eu dans la rubrique téléchargement,
- Page 47 : les missions des centres de référence,
- Page 49 : la vie au quotidien – absence de régime, le suivi médical…,
- Pages suivantes : la vie en société, à l’école, au travail, les sports…, le milieu professionnel,
- Page 87 : les sources d’informations, dont EURORDIS, -communauté des CAPS.
- NOMIDLLIANCE présidé par notre amie KAREN de San Francisco www.nomidalliance.net
- Page 91 : présentation de votre association,
- Page 92 : le glossaire.